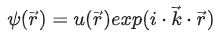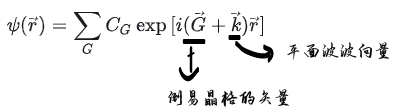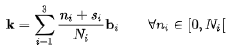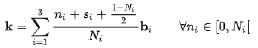现在需要对第一布里渊区进行分析,使用的方法叫做K点采样 K点采样中的K点是布里渊区中的特定点,而不是特定的倒格子密度 是用来充分描述能带结构和电子性质 K点采样通常会按照布里渊区的原胞轴向进行设置采样点 根据Bloch定理,单电子波函数可以由一个具有晶格周期的函数调制的平面波表示
由于平面波是一个标准正交基组,因此任何函数都可以由一个平面波基组表示。
周期性函数代入波函数可以得到,下面这个式子
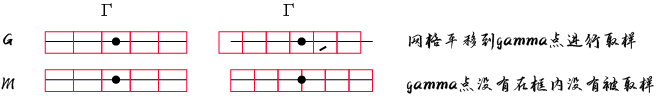
平面波波向量 k又称“布洛赫波向量”,它与约化普朗克常数的乘积即为粒子的晶格动量 它可以表征不同原胞间电子波函数的位相变化 其大小只在一个倒易点阵向量之内才与波函数满足一一对应关系 所以通常只考虑第一布里渊区内的波向量,即所谓“简约波向量” 对一个给定的波矢和势场分布 电子运动的薛定谔方程具有一系列解,称为电子的能带 常用波函数的下标n 以区别 这些能带的能量在k的各个单值区分界处存在有限大小的空隙,称为能隙 在第一布里渊区中所有能量本征态的集合构成了电子的能带结构 在单电子近似的框架内,如一个周期性势场具有离散的平移对称性 电子的波函数可以由能带和晶格动量来共同标记 电子运动的宏观性质都可以根据能带结构及相应的波函数来可靠地预言。 上述结果的一个推论为:在确定的完整晶体结构中 布洛赫波向量k是一个守恒量(以倒易点阵向量为模) 布洛赫电子的运动具有波矢量k,它描述了电子在晶体内的运动方向和速度 在完整晶体中,我们可以将电子的运动分解为一系列在倒空间中的波矢量的叠加 所以该模型又称为近自由电子近似 导体的电阻仅仅来自那些破坏了势场周期性的晶体缺陷以及电子与声子的相互作用 K点采样的常规方法有2种 选择是以Γ为中心的(G,g)或Monkhorst-Pack方案(M,m) Γ心网格,以下 k 点对布里渊区进行采样
Monkhorst-Pack 网状物,k 点网格由此定义产生
对于两个网格,点之间的间距相同。 唯一的区别是转变(1-Ni)/2 在 Monkhorst-Pack 网格的分子中 对于奇数个细分,该项是一个整数,因此由于周期边界,两个网格一致 当细分数为偶数时,以Γ为中心的网格与 Monkhorst-Pack 相比将移动(1-Ni)/2
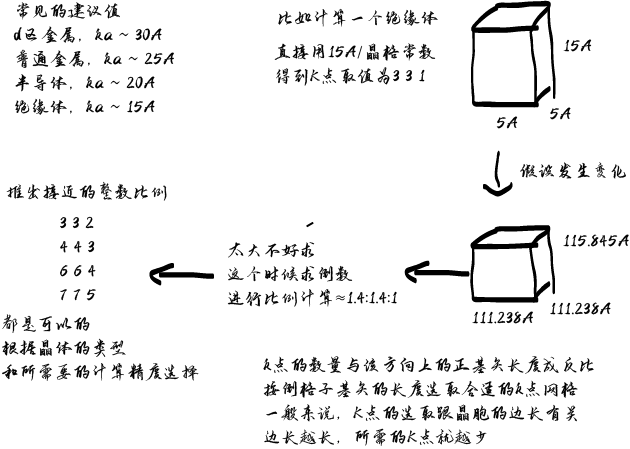
Gamma撒点在MP撒点的基础上进行平移,保证采到Gamma点 MP方法的核心是将布里渊区内的积分分解成N1×N2×N3个网格点上的离散积分 这里N1、N2和N3分别是积分在每个布里渊区方向上的网格点数 k点之间的间距被选择为尽可能小,同时仍然允许模拟收敛 这是因为k点之间较小的间距可以更好地描述晶体的电子结构 尤其是对于具有强电子相关性的复杂材料 然而,对于一些材料,特别是那些带隙小或电子相关性强的材料 k点之间的间距会对模拟结果产生重大影响 在这些情况下,有必要使用非常精细的网格来准确地模拟材料的电子结构 对于k点的需求,金属>>半导体,绝缘体 不过呢,很多时候主要还是受硬件限制简约化可以使k点的数目大大下降 对于原子数较多的体系的计算,就需要谨慎的尝试k点数目 在避免或者预先评估环绕误差的前提下尽量减少k点数目。 一般计算要在第一布里渊区均匀撒点,能带计算在高对称点连线路径上取值 *只有在计算简单体系/超大体系只考虑选择计算gamma点 只计算Gamma点可能会在某些情况下导致结果的不准确 超大体系可以的原因是因为对应的布里渊区成反比 晶胞越大,布里渊区越小,所以太大可以只选择gamma点
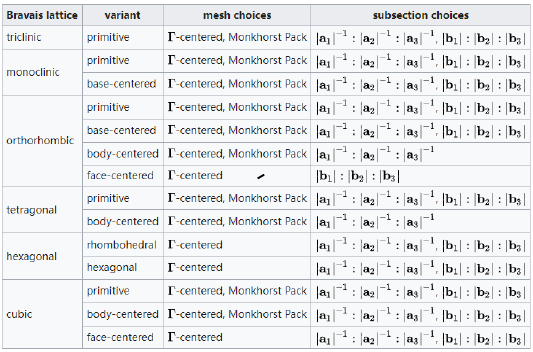
请参阅下表,根据系统的对称性做出明智的选择
k点和体系的晶格是具有关联性的 所以晶格的对称性影响k点的对称性 最终影响取值 又因为M网格的收敛速度可能比G的网格更快 所以我们先考虑M特殊体系再考虑G gamma点的选取是必须的,所以G方法都可以使用 计算一个分子或者单原子时,总是选取一个Gamma点 因为这个点上的能级分布基本反映了整个能带 每个能带都和k相关性不大;而对于半导体则需要多一点的k点 而金属需要最多的k点,因为金属中的电子的非局域性很强 |
Powered by Discuz! X3.5
© 2001-2024 Discuz! Team.