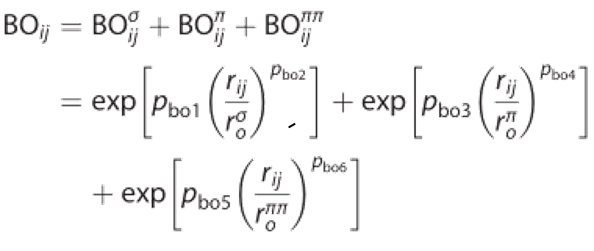势能分为依赖键和不依赖键的贡献。键序由原子间距离直接计算,使用经验公式:
其中BO为原子i和j之间的键序,rij为原子间距离,ro为平衡键长,pbo为经验参数。方程是连续的,通过σ、π和π键特征之间的转换不存在不连续。这就产生了一个可微的势能面,这是计算原子间作用力所必需的。该键序公式适用于过渡态结构中长距离共价相互作用的特点,使力场能够准确预测反应势垒。这个共价范围通常被认为是5埃,这足以让大多数元素捕捉到最弱的共价相互作用,但可以扩展到这个范围之外;对于共价半径非常大的元素,有时可能需要这样做。然而,这种远距离共价键的特征需要添加键序校正来消除非键邻居之间的虚假键特征,例如甲烷分子中相邻的H原子。电势中依赖于键序的项,如键能和角应变,直接从修正后的键序计算。最后,在每次迭代中应用电荷平衡方案来计算原子电荷分布,然后将其用于计算库仑相互作用。 请注意,ReaxFF中的非键和键项是独立计算的——基于键序的项和与范德华和库仑相关的项之间没有信息传递。对于所有材料和分子,基于键序的项和非键的项都被计算,使ReaxFF能够应用于主要的共价和离子材料,而无需用户输入。 ReaxFF立场首先根据两个原子间间距以及计算出BO,再根据BO计算出共价相互作用(键能,键角能,二面角能以及corrections等)。当某个时间步的某个键的BO小于某个数值(默认值是0.3),会判断该化学键断裂从而导致共价相互作用消失。反之,如果在某时间步的某个两个原子间的BO超过0.3,会判断这两个原子间生成化学键,从而产生相互作用。(这个步骤与LAMMPS的fix bond/react命令相似)。 |
Powered by Discuz! X3.5
© 2001-2024 Discuz! Team.