ReaxFF训练过程中的参数优化 在FF训练步骤中优化的参数列于表S1。表S2列出了我们进行FF优化的含Mo-S化合物和凝聚相周期结构。 量化计算方法处理分子: B3LYP交换相关泛函和LAV3P**基组 第一性原理计算放法处理周期性结构: GGA-PBE PAW方法 Γ-centered Monkhorst - Pack网格 平方波能量截止值为400 eV 作用力收敛值0.02 eV/Å
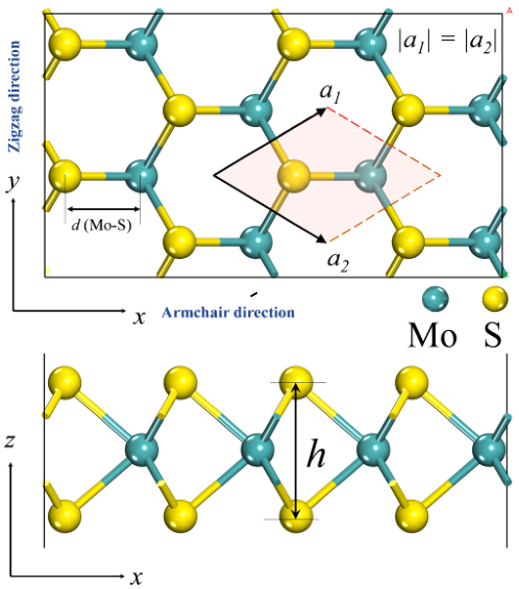
通过将相邻两层中的Mo原子限制在c轴距离d内,只允许S原子弛豫,可以实现双层MoS2在不同层间距离d处的结构弛豫。在所有的双层结构中都保留了AA的堆叠(当沿着c轴观察时相邻层中的Mo和S相互遮蔽)。采用微推弹性带(NEB)法计算激活势垒。尽管存在标准DFT固有的带隙低估问题,但在没有电子激发的情况下,该误差不应影响本研究中讨论的结构能。 二硫化钼的ReaxFF势能。ReaxFF中的能量被写成键能(Ebond)、键角(Eval)、扭转角能量(Etor)、孤对电子项(Elp)、过配位的能量矫正项(Eover)和欠配位的能量矫正项(Eunder)能量惩罚加上非键范德瓦尔斯(Evdw)和库仑(ECoulombic)贡献的总和 Esystem=Ebond+Eval+Etor+Eover+Eunder+Elp+EvdW+Ecoulombic 在力场训练步骤中被优化的参数列表 键解离和键角畸形变的DFT和ReaxFF计算 Mo−S键裂解和形成的计算用于力场优化 ReaxFF与DFT能量对比案例 室温下原始2H MoS2薄片的单轴应力-应变响应 空位迁移能演化及优化后的ReaxFF反应力场参数 初始配置用于优化BGF格式的潜力 trainset.in: 力场训练集
|
Powered by Discuz! X3.5
© 2001-2024 Discuz! Team.