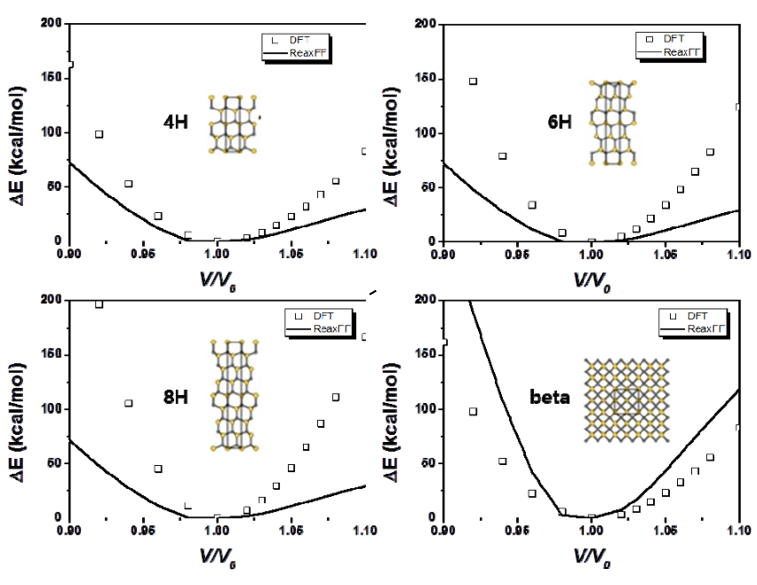
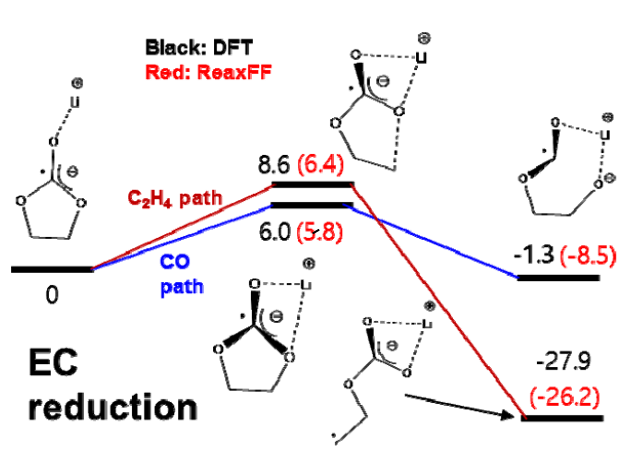
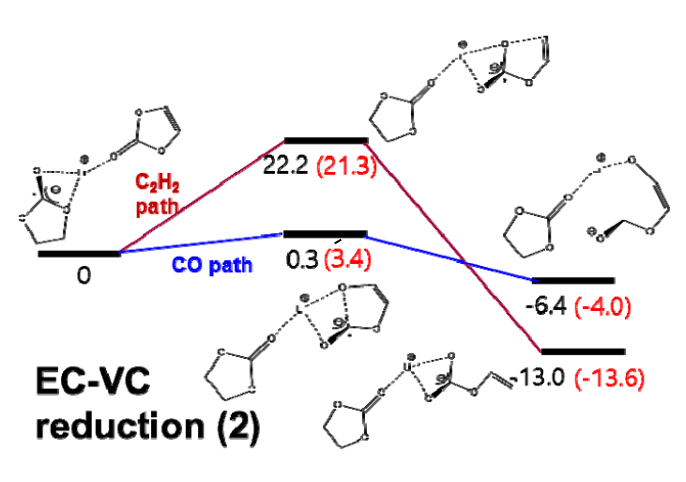
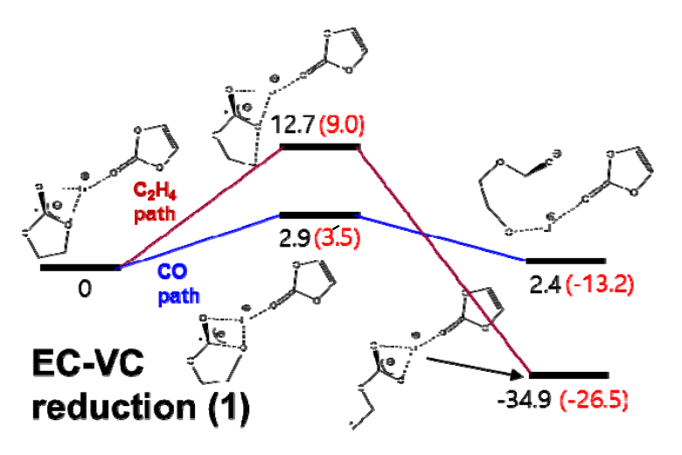
为了模拟Si和SiOx阳极上SEI形成行为随电解质类型和成分的变化,我们使用了反应力场(ReaxFF)进行MD模拟。本次计算涉及到Si-Li-O-C-H-F力场参数,由于作者之前已经开发了Si-Li-O力场参数,C-H和O-H参数可以查找参考文献,因此本次开发Si-C, Li-C, O-C, Si-H, Li-H, F-F, C-F, H-F, Li-F, 和Si-F的键,键角,扭转角相关参数。 根据密度泛函理论(DFT)计算得到的训练集对各种分子和晶体的ReaxFF参数进行优化。为了计算不同分子结构的键解离能、键角弯曲能和扭转能,我们在Q-Chem软件中使用了B3LYP杂化泛函和6-311G**基组。此外,我们还考虑了碳酸乙烯(EC)分子和碳酸乙烯(ECVC)配合物的DFT还原途径,以优化ReaxFF。此外,我们使用平面波基组处理周期结构,计算了几种SiC晶体的状态方程(EOSs)。使用VASP软件在PBE泛函进行计算,以及使用投影缀加波贋势。我们使用了400 eV的动能截止能量和Monkhorst-Pack方案来生成k空间网格。 拟合处理需要DFT与ReaxFF结果进行逐一比对
作者使用ReaxFF力场进行MD模拟研究Si和SiOx阳极上的SEI形成行为以及关于电解液各成分变化的关系。 标准的ReaxFF不包括显式电子效应,尽管最近开发的eReaxFF有。因此,在研究ReaxFF参数时,只考虑单电子在电解液中Li+与其他物质的还原机理,换句话说,在这项工作中,当锂原子与电解质和添加剂分子相互作用时,它们之间的化学反应被描述为电子自动参与反应。利用这种方法,我们可以间接地描述Li——电解质配合物的还原反应。 DFT与ReaxFF在Li+-EC配合物单电子还原中的比较 Li+EC- vc配合物单电子还原(EC解离)的DFT和ReaxFF比较 Li+EC-VC配合物单电子还原(VC解离)的DFT和ReaxFF比较
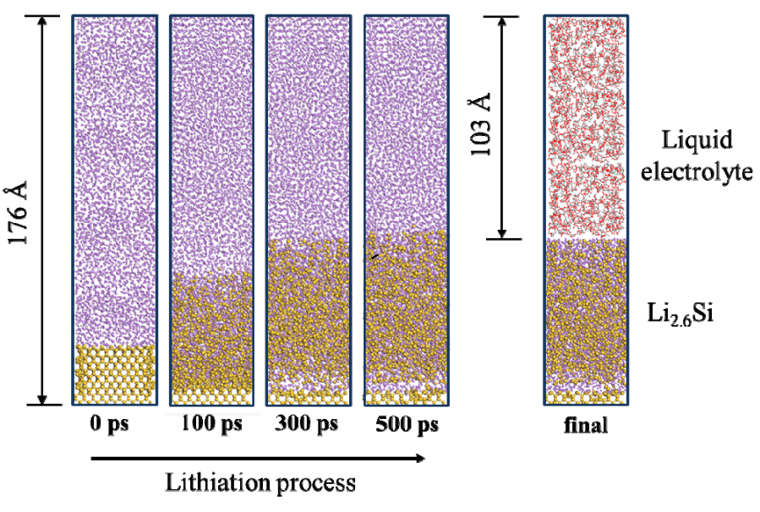
作为模拟模型,我们考虑了包括锂化Si基阳极之间界面的结构,以及锂化Si基阳极和硅基阳极之间的界面,但它可能会导致锂化Si基阳极的结构。在ReaxFF-MD模拟的基础上,构建了LixSi和LixSiOx两种不同的结构。在模拟单元34.6 Å × 34.6 Å x 176 Å,我们最初放置了一个Si(100)切面,共18层和Li原子,其中Si和Li原子的数量分别为1634和8000。 使用ReaxFF,我们在正则系综条件下进行了500 ps的MD模拟,导致Si的锂化。在MD模拟后,我们移除了所有不与Si原子结合的Li原子,产生真空区域。然后,在真空区域,我们放入电解液。最终模型中包含Li2.6Si阳极和电解质的原子总数约为16000 ~ 17000。 Si锂化的界面是计算跑出来的,不是直接建模得到的。这里是为后续做的准备。
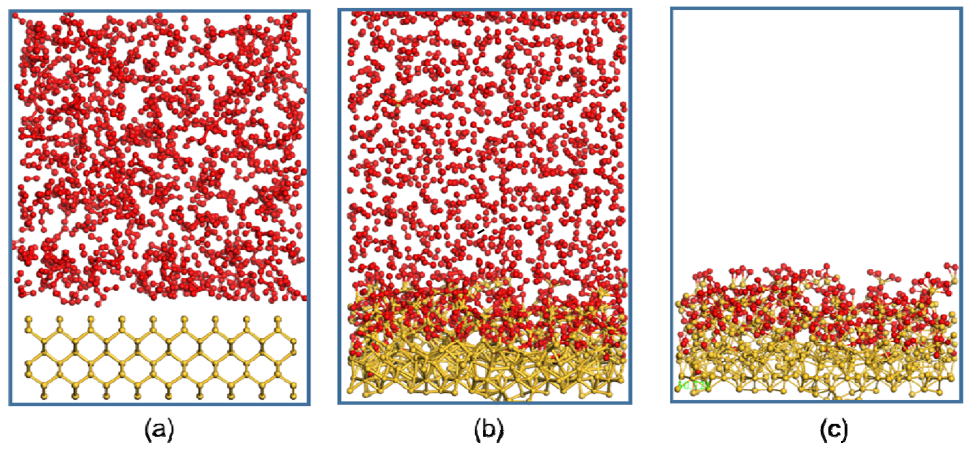
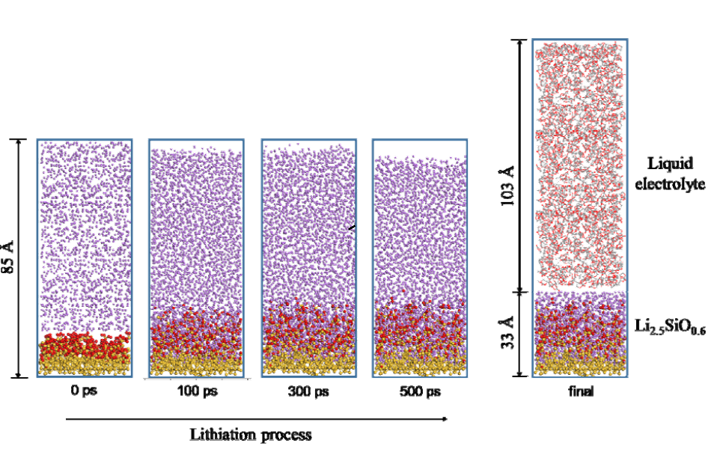
获得SiOx,我们使用ReaxFF-MD进行处理,建模尺寸设置为34.6 Å × 34.6 Å x 49Å),这里使用Si(100)切面和O2,温度为1000 K,经过500 ps的MD模拟,我们得到了SiO0.6的组成。在此基础上,通过对Li2.5SiO0.6阳极和液态电解质的模拟,得到了Li2.5SiO0.6阳极和液态电解质的模拟模型。最终模型中包含Li2.5SiO0.6阳极和电解质的原子总数约为13,000 ~ 14,000。 这步同样是用来建模的步骤。
|
Powered by Discuz! X3.5
© 2001-2024 Discuz! Team.