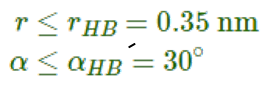gmx hbond [-f [<.xtc/.trr/...>]] [-s [<.tpr>]] [-n [<.ndx>]] [-num [<.xvg>]] [-g [<.log>]] [-ac [<.xvg>]] [-dist [<.xvg>]] [-ang [<.xvg>]] [-hx [<.xvg>]] [-hbn [<.ndx>]] [-hbm [<.xpm>]] [-don [<.xvg>]] [-dan [<.xvg>]] [-life [<.xvg>]] [-nhbdist [<.xvg>]] [-b <time>] [-e <time>] [-dt <time>] [-tu <enum>] [-xvg <enum>] [-a <real>] [-r <real>] [-[no]da] [-r2 <real>] [-abin <real>] [-rbin <real>] [-[no]nitacc] [-[no]contact] [-shell <real>] [-fitstart <real>] [-fitend <real>] [-temp <real>] [-dump <int>] [-max_hb <real>] [-[no]merge] [-acflen <int>] [-[no]normalize] [-P <enum>] [-fitfn <enum>] [-beginfit <real>] [-endfit <real>] gmx-hbond计算和分析氢键。氢键是基于氢-供体-受体的角度(零被扩展)和供体-受体(或使用-noda的氢-受体)的距离的截止来确定的。OH和NH基团被视为供体,O始终是受体,默认情况下N是受体,但这可以使用-nitacc进行切换。假定虚设氢原子连接到前面的第一个非氢原子。 您需要指定两个组进行分析,这两个组必须相同或不重叠。分析了两个基团之间的所有氢键。 如果您设置了-shell,您将被要求提供一个额外的索引组,该索引组应该只包含一个原子。在这种情况下,只考虑与一个原子相距壳层距离内的原子之间的氢键。 通过选项-ac,氢键的速率常数可以用Luzar和Chandler的模型导出(Nature 379:551996;J.Chem.Phys.113:232000)。如果通过使用-contact选项来分析接触动力学,则n(t)可以被定义为在时间t不在接触距离r内的所有对(对应于将-r2选项保留在默认值0)或在距离r2内的所有配对(对应于使用选项-r2设置第二截止值)。有关更多详细信息和定义,请参阅上述文献。 gmx hbond程序用于分析所有可能的施体D和受体A之间的 氢键 (HB). 分析时使用几何准则决定氢键的存在与否
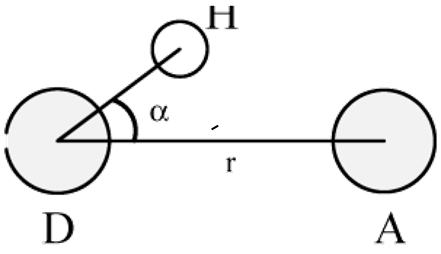
参考值 rHB=0.35nm,对应于SPC水模型RDF的第一极小位置(参看图 8.3). gmx hbond程序以下面的方式分析两组原子(它们必须相同或没有重叠)或指定的施体-氢-受体之间所有可能存在的氢键:
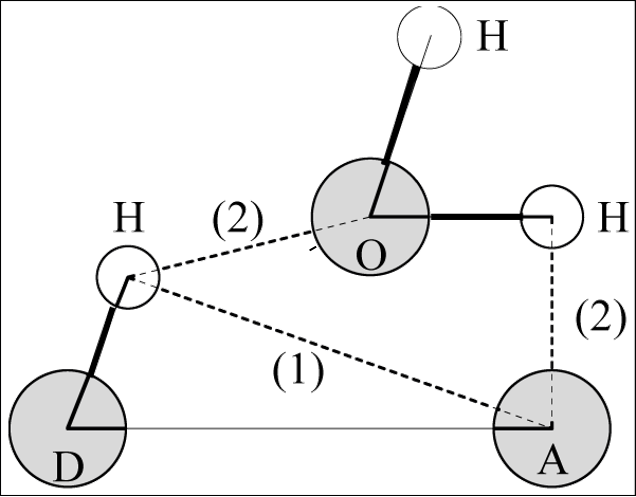
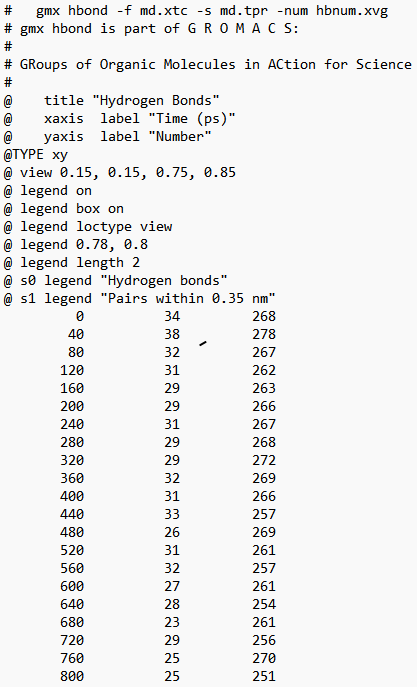
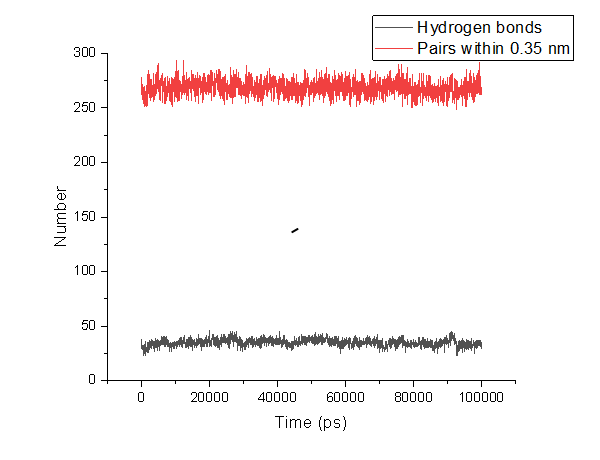
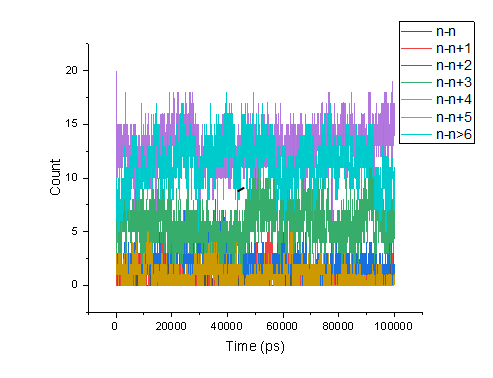
水对氢键的插入. (1) 两残基间正常的氢键. (2) 通过一个水分子形成的氢键桥. -num:作为时间函数的氢键数目Time~Num。 -ac:所有氢键的存在函数(0或1)的所有自相关的平均值。 -dist:所有氢键的距离分布。 -ang:所有氢键的角度分布。 -hx:作为时间函数的n-n+i氢键的数量,其中n和n+i代表残基数量,i的范围为0至6。这包括与蛋白质中的螺旋相关的n-n+3、n-n+4和n-n+5氢键。 -hbn:所有选定的基团,所选基团的供体、氢和受体,所有基团的所有氢键原子和所有参与插入的溶剂原子。 -hbm:所有框架上所有氢键的存在矩阵,它还包含溶剂插入氢键的信息。排序与-hbn索引文件中的排序相同。 -dan:写下每个时间段分析的供体和受体的数量。这在使用-shell时特别有用。 -nhbdist:计算每个氢的HBonds数量,以便将结果与拉曼光谱进行比较。 注意:选项-ac、-life、-hbn和-hbm需要的内存量与所选组中供体总数乘以受体总数成比例。 指定输入文件的选项: -f[<.xtc/.tr/…>](traj.xtc) 轨迹:xtc trr cpt gro g96 pdb tng -s[<.tpr>](拓扑.tpr) 可移植的xdr运行输入文件 -n[<.ndx>](索引.ndx)(可选) 索引文件 指定输出文件的选项: -num[<.xvg>](hbnum.xvg) xvgr/xmgr文件 -g[<.log>](hbond.log)(可选) 日志文件 -ac[<.xvg>](hbac.xvg)(可选) xvgr/xmgr文件 -dist[<.xvg>](hbdist.xvg)(可选) xvgr/xmgr文件 -ang[<.xvg>](hbang.xvg)(可选) xvgr/xmgr文件 -hx[<.xvg>](hbhelix.xvg)(可选) xvgr/xmgr文件 -hbn[<.ndx>](hbond.ndx)(可选) 索引文件 -hbm[<.xpm>](hbmap.xpm)(可选) X PixMap兼容矩阵文件 -don[<.xvg>](donor.xvg)(可选) xvgr/xmgr文件 -dan[<.xvg>](danum.xvg)(可选) xvgr/xmgr文件 -life[<.xvg>](hblife.xvg)(可选) xvgr/xmgr文件 -nhbdist[<.xvg>](nhbdist.xvg)(可选) xvgr/xmgr文件 其他选项: -b<time>(0) 从轨迹读取第一帧的时间(默认单位为ps) -e<time>(0) 从轨迹读取的最后一帧的时间(默认单位为ps) -dt<time>(0) 仅当t MOD dt=第一次时使用帧(默认单位ps) -tu<enum>(ps) 时间值的单位:fs、ps、ns、us、ms、s -xvg<enum>(xmgrace) xvg绘图格式:xmgrace、xmgr、none -a<real>(30) 截止角(角度,氢-供体-受体) -r<real>(0.35) 截止半径(nm,X-受体,见下一个选项) -[no]da (yes) 使用距离供体受体(如果为TRUE)或氢受体(FALSE) -r2<real>(0) 第二个截止半径。主要用于-contact和-ac -abin<real>(1) 角度分布间隔宽度(角度) -rbin<real>(0.005) 距离分布间隔宽度(nm) -[no]nitacc(yes) 将N原子视为受体 -[no]contact (no) 不要寻找氢键,只需寻找截止距离内的触点 -shell<real>(-1) 当>0时,仅计算一个粒子周围#nm壳层内的氢键 -fitstart<real>(1) 开始拟合相关函数的时间(ps),以便获得HB断裂和形成的正向和反向速率常数。使用-gemfit,我们建议-fitstart 0 -fitend<real>(60) 停止拟合相关函数以获得HB断裂和形成的正向和反向速率常数的时间(ps)(仅使用-gemfit) -temp <real> (298.15) 用于计算对应于HB断裂和重整的吉布斯能的温度(K) -dump <int> (0) 将前N个氢键ACF转储到单个.xvg文件中进行调试 -max_hb<real>(0) 用于归一化HB自相关函数的理论最大氢键数。在程序估计错误的情况下可能有用 -[no]merge (yes) 同一供体和受体之间但具有不同氢的氢键被视为单个氢键。主要对ACF很重要。 -nthreads<int>(0) 自相关上用于并行循环的线程数。nThreads<=0表示最大线程数。需要与OpenMP链接。线程数量受内核数量(OpenMP v.3之前)或环境变量OMP_THREAD_LIMIT(OpenMP v.3)的限制 -acflen<int>(-1) ACF的长度,默认为帧数的一半 -[no]normalize (yes) 标准化ACF -P<enum>(0) ACF的勒让德多项式阶数(0表示无):0,1,2,3 -fitfn<enum>(无) 拟合函数:none、exp、aexp、exp_exp、exp5、exp7、exp9 -beginfit<real>(0) 开始相关函数的指数拟合的时间 -endfit <real> (-1) 结束相关函数的指数拟合的时间,-1是直到结束 gmx hbond -f md.xtc -s md.tpr -num hbnum.xvg -g hbond.log -ac hbac.xvg -dist hbdist.xvg -ang hbang.xvg -hx hbhelix.xvg -hbn hbond.ndx -hbm hbmap.xpm -don donor.xvg -dan danum.xvg -life hblife.xvg -nhbdist nhbdist.xvg 全部执行会发现很多东西无法进行执行 gmx hbond -f md.xtc -s md.tpr -num hbnum.xvg
gmx hbond -f md.xtc -s md.tpr -g hbond.log(没有生成hbond.log,而是生成hbnum.xvg) gmx hbond -f md.xtc -s md.tpr -ac hbac.xvg(还是会生成hbnum.xvg) gmx hbond -f md.xtc -s md.tpr -dist hbdist.xvg
gmx hbond -f md.xtc -s md.tpr -ang hbang.xvg(还是会生成hbnum.xvg) gmx hbond -f md.xtc -s md.tpr -hx hbhelix.xvg
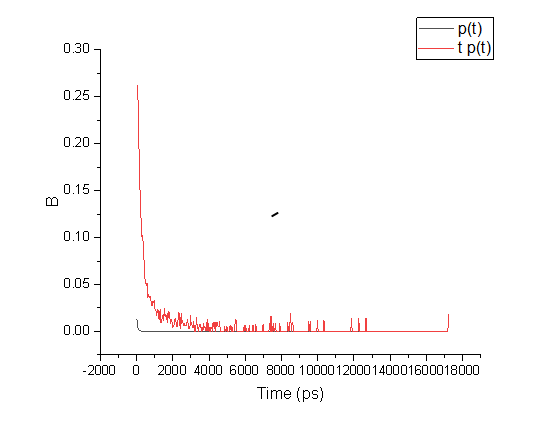
gmx hbond -f md.xtc -s md.tpr -hbm hbmap.xpm gmx xpm2ps -f hbmap.xpm -o hbmap.eps -rainbow blue gmx hbond -f md.xtc -s md.tpr -life hblife.xvg
第一列(横坐标)是时间 第二列大抵是氢键能连续保持这么长时间不断掉的几率 第三列是前两列相乘 gmx hbond -f md.xtc -s md.tpr -da yes -num hbnum1.xvg gmx hbond -f md.xtc -s md.tpr -da no -num hbnum2.xvg 使用距离分析供体-受体(yes)或氢-受体(no) gmx hbond -f md.xtc -s md.tpr -nitacc yes -num hbnum1.xvg gmx hbond -f md.xtc -s md.tpr -nitacc no -num hbnum2.xvg 如果N不作为受体肯定会减少 |
Powered by Discuz! X3.5
© 2001-2024 Discuz! Team.