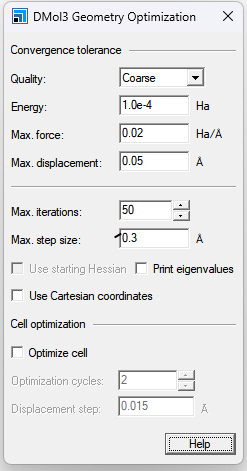
Quality:为优化循环之间的能量变化、最大力和最大位移设置几何优化收敛阈值。当满足能量收敛以及位移或梯度标准时,优化将停止。如果计算的初始梯度低于阈值,则优化将成功停止,无需执行任何步骤,也无需比较位移和能量。或者,可以独立指定“Energy”、“Max. force”和“Max. displacement”的阈值。如果为这些设置中的任何一个输入自己的值,则“Quality”在“DMol3 Geometry Optimization”对话框和DMol3计算对话框的“Setup”选项卡上都显示为“Customized”。对于具有B3LYP或HF函数的分子系统的计算,用户可能必须在.input文件中明确增加B3LYP-或HF比积分网格密度。
Energy:在几何优化过程中,以Hartree为单位指定最大能量变化的收敛阈值。 Max. force:指定几何优化过程中最大力的收敛阈值,单位为HartreeÅ-1。 Max. displacement:指定几何优化过程中最大位移的收敛阈值,单位为Å。 Max. iterations:指定几何体优化循环的最大次数。如果达到这个循环数,则即使不满足收敛标准,计算也将停止。这些收敛公差和最大迭代设置仅适用于单位单元中的原子坐标优化。 Max. step size:指定任何笛卡尔坐标允许的最大变化。几何位移被截断,使其小于该值。这可以防止优化器采取不合理的步骤。DMol3中的默认优化器使用离域内部坐标,最大笛卡尔步长不直接适用于此方法。如果您观察到最小化过程中的实际位移过大并导致较大的能量变化,则应减小“最大步长”的值。 Use starting Hessian:选中时,与当前模型关联的Hessian将用作新计算中的初始Hessian。如果未选中,最小化将在没有Hessian的情况下开始。您可以从多个来源获得起始Hessian,如导入Hessian文件中所述。当对称性被激活时,不可能在DMol3中使用起始Hessian进行几何优化。 Print eigenvalues:检查时,在每个收敛SCF循环结束时打印Kohn Sham特征值。 Use Cartesian coordinates:选中时,笛卡尔坐标用于几何优化。 Optimize cell:选中时,除了原子坐标外,还会在几何优化过程中优化单元参数。默认设置为未选中。 只有当当前活动文档包含周期性结构时,才启用此选项。如果禁用或未选中此选项,则“优化周期”和“置换步骤”不可用。 晶胞优化需要晶胞矢量的许多小变化和每个位移的局部优化。 如果选择了“优化单元和对称性”,则无法在同一运行中计算振动。 Optimization cycles:确定单元优化步骤的数量。 DMol3运行所有指定的单元优化步骤。从第二个周期开始,信息将打印到.outtl文件中,用户可以评估单元优化的进度。 Displacement step::单元矢量位移的大小。 |
Powered by Discuz! X3.5
© 2001-2024 Discuz! Team.