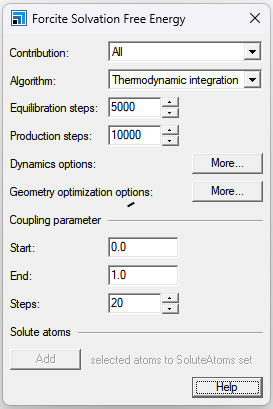
Contribution: 指定要计算的自由能量贡献。指定“全部”将在一次计算中计算所有贡献。 Algorithm: 指定用于计算溶剂化自由能的算法。 Equilibration steps:平衡步数,默认值=5000 Production steps: 采样步数,默认值=10000 Dynamics options:,该对话框允许您指定在计算的平衡和生产阶段使用的动力学选项。 Geometry optimization options:该对话框允许您指定平衡运行之前的几何优化阶段的详细信息。 Coupling parameter:指定耦合参数的间隔以及该间隔中的步数。相反的过程可以通过指定从1到0的耦合参数来实现。 Start: 默认值为0.0。 End: 默认值为1.0。 Steps: 默认值=20。 对于反向过程,所报告的自由能的符号被反转,从而与正向过程的符号相匹配。 Solute atoms:将选定的原子添加到SoluteAtoms集合:使用活动的3D原子文档中当前选定的原子创建一组名为SoluteAtomics的原子。如果已经为活动文档定义了名为SoluteAtoms的集合,则任何尚未成为该集合成员的选定原子都将添加到该集合中。
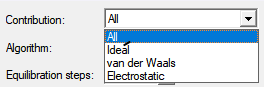
将在一次计算中计算所有贡献 All (default):全部(默认) Ideal:理论值 van der Waals:范德华力 Electrostatic:静电力
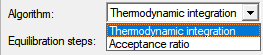
Thermodynamic integration:TI热力学积分 Acceptance ratio:Bennett接受比例法 自由能微扰法:Free Energy Perturbation (FEP) 热力学积分法:Thermodynamic Integration (TI) Bennett接受比例法:Bennett Acceptance Ratio (BAR) 多态Bennett接受比例法:Multistate Bennett Acceptance Ratio (MBAR) 关于这几种自由能计算方法,有几条建议: 当所有能量差都可以得到时,推荐使用MBAR方法。可以证明,在多态模拟时,该方法可以给出最小误差估计; BAR方法是MBAR方法的一种特例。在λ中间态重叠性足够好时,BAR可以给出与MBAR一样好的结果; 在中间态窗口足够多的时候,TI方法通常可以给出与MBAR方法一致的结果。但是其积分误差很难预先估计; WHAM方法是MBAR方法的近似,没有令人信服的理由证明选用WHAM方法而不用会更好的MBAR方法。 |
Powered by Discuz! X3.5
© 2001-2024 Discuz! Team.