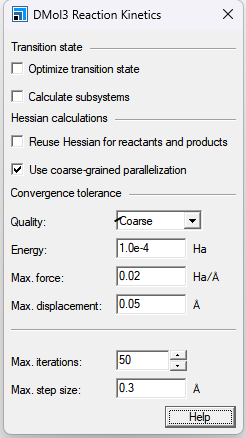
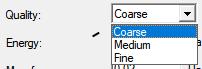
Optimize transition state:在计算过渡状态的Hessian和总能量之前,进行过渡状态优化。如果过渡状态的Hessian源于几何优化,则它可能是迭代更新的近似值。在这种情况下,它可能不包含任何虚频率,即使结构非常接近实际的过渡状态。为了避免这种情况,请确保在开始反应动力学计算之前进行振动分析。 DMol3只能在过渡状态的Hessian可用并且具有单个虚频率的情况下执行过渡状态优化。 重要!若要模拟具有非零电荷的缔合或离解反应,必须为每个子系统手动指定电荷。 在此DMol3反应动力学对话框中将电荷指定为零,并保存输入文件。 打开每个输入文件并指定适用于每个系统的电荷。 使用DMol3作业文件对话框为系统运行反应动力学模拟。 Calculate Subsystems::检查时,每个反应成分都是单独计算的。 这仅适用于表面反应。 Reuse Hessian for reactants and products:检查时,如果任何反应物或产品结构已经导入了Hessian,则不会对该结构进行额外的几何优化或Hessian计算。 Use coarse-grained parallelization:选中时,数值位移频率计算使用粗粒度并行化。生成系统Hessian矩阵所需的几何位移计算在所有可用的计算节点上均匀分布。然后以串行模式运行每个位移,并在计算结束时收集最终的Hessian元素。对于具有许多振动模式和非平凡数量的计算节点的较大系统,这是优选的方法。 如果未检查,则按顺序计算数值位移,每个位移都是在并行DMol3过程中计算的。这对于具有较少正常模式的较小系统或具有有限数量计算节点的机器来说可能是有利的。 默认情况下,使用分析力的两点差来评估Hessian。您可以通过修改输入文件中的Vibration_Steps关键字来更改此设置。有关如何修改输入文件的信息,请参阅DMol3作业文件主题。 Quality:定义优化循环之间的能量变化、最大力和最大位移的几何体和过渡状态优化收敛阈值。当满足能量收敛以及位移或梯度标准时,优化停止。如果计算的初始梯度低于阈值,则优化成功停止,无需执行任何步骤,也无需比较位移和能量。 Energy:在几何优化过程中,以Hartree为单位指定最大能量变化的收敛阈值。 Max. force:指定几何优化过程中最大力的收敛阈值,单位为HartreeÅ-1。 Max. displacement:指定几何优化过程中最大位移的收敛阈值,单位为Å。 Max. iterations:指定几何体优化循环的最大次数。如果达到这个循环数,则即使不满足收敛标准,计算也会停止。 Max. step size:指定任何笛卡尔坐标允许的最大变化。 几何位移被截断,使其小于该值。这可以防止优化器采取不合理的步骤。 DMol3中的默认优化器使用离域内部坐标,最大笛卡尔步长不直接适用于此方法。 如果最小化过程中的实际位移过大并导致较大的能量变化,请减小“最大步长”的值。
|
Powered by Discuz! X3.5
© 2001-2024 Discuz! Team.