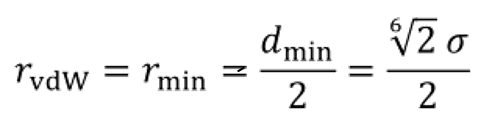角度和二面角影响CG模型的质量,因为它们控制着分子的构象灵活性。在Martini 3中,键距同样重要,因为它们将定义CG模型的体积、形状和头密度,这对力场的整体行为有重要影响。在这里,我们首先描述了获得合理好的模型的一般规则,然后描述了基于体积形状的细化。 几何中心映射:一般来说,从质心(COM)映射的原子结构获得的键距证明是不令人满意的,因为它们经常导致过高的填充密度。这是由于忽略了CG模型(例如,苯的三角形与六边形)的较低分辨率而导致的几何不匹配不考虑氢原子的体积。相反,使用基于几何中心(COG)的原子结构映射,将氢原子考虑在内,可以更好地再现分子体积和质量密度。因此,由Cog映射的原子结构获得的键距构成了在Martini 3中获得键参数的标准程序。当映射更接近原子分辨率时,即使用T-beads时,COG映射尤为重要。在4-1映射的烷烃链中,基于COM和基于COG的键距确实几乎相等。相比之下,它们在2比1映射环的情况下差别很大。例如,苯的COM键长为0.21 nm,而COG键长为0.29 nm。基于cog和基于COM的键距可以直接从原子结构中提取。因此,从COG映射的原子结构中提取的键距应该是自动参数化方案的标准,例如Auto-Martini41或PyCGtool47算法。 基于体积和形状的改进:考虑到Martini模型的CG分辨率,仅仅遵循基于COG的映射方案,无法在所有情况下获得完美匹配的密度或包装。因此,如果需要更高的精度,键距可以进一步微调。为此,溶剂可及表面积(SASA)、分子体积和形状可以作为优化键距的指标。 键合参数、力常数和排除:在Martini 3中,连接珠子之间的非键合相互作用总是被排除,无论是在默认键合项中还是在蛋白质的弹性网络中。正如Alessandri等人所证明的,在这种情况下,应该避免弱的结合力常数(低于700 kJ/mol),因为它们可能会使分子内的CG珠靠得太近,导致与环境的相互作用过高,并造成伪影(例如,粒子彼此重叠,过度聚集)。排除也可以应用于第二或第三邻居,以帮助在某些情况下的角度和二面角分布的表示。对于环,甚至使用更高阶的不相容,这可以帮助保持它们的构象数值稳定。特别是,非虚拟粒子(见第C4节)和用于构建它们的参考珠子之间应该始终使用排斥。 要计算SASA,可以使用gromacs工具gmx SASA。对于原子参考,我们推荐Rowland和Taylor的范德华半径(vdW)。对于Martini CG分子,R-、S-和t-珠子的vdW半径分别为0.264、0.230和0.191nm。这些计算为LJ势函数最小值的位置,给定各种珠子的LJ的σ值:
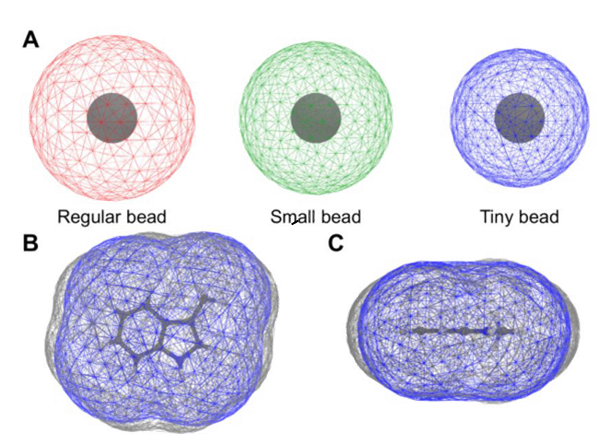
其中:mol.pdb(或mol.gro或trajectory.xtc)是包含分子几何结构的文件;sasa.XVG是输出文件;-probe指定探针的大小,我们为其选择了T珠子大小。探针的大小不一定是这个,但在比较不同分子和模型的SASA值时,包括原子和CG结果的比较,必须使用相同的探针大小。指定SASA计算精度的-ndots标志应该设置为至少4800点,以获得准确的SASA值。 对于刚性分子,如小芳香环,SASA值已经计算了能量最小化的AA和CG几何形状。对于更灵活的分子,可以比较沿AA和CG轨迹的SASA值的分布。最后,还可以生成Connolly表面(surf.pdb),以便能够在3D中可视化分子体积。这可以用下面的命令来完成: gmx sasa -s mol.pdb -o sasa.xvg -probe 0.191 -ndots 240 -q surf.pdb 请注意,为了生成surf.pdb文件更容易被VMD等可视化程序处理,用于计算的点数(-ndots)可以减少到,例如240。这样的表面可以计算AA和CG模型,如补充图7BC中的3-甲基吲哚分子。排列的表面有助于确定键距,可以根据AA和CG表面之间的差异进行优化。在http://cgmartini.nl/上提供了一个教程,其中包含更多最新的细节和示例。
|
Powered by Discuz! X3.5
© 2001-2024 Discuz! Team.