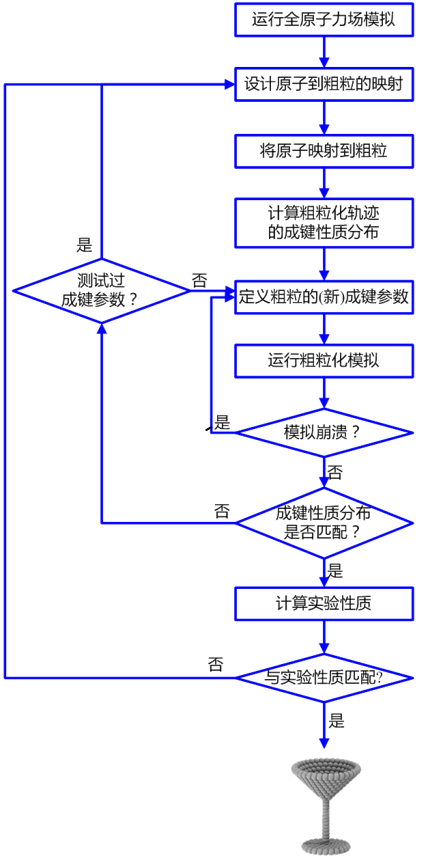
对于最可靠的参数化,原子模拟溶液中的分子。Martini主要以Gromos为基础,但你可以使用任何你喜欢的力场。如果不存在拓扑结构,您可以使用ATB或GAFF力场来构建一个拓扑结构(这可能也是相当多的工作)。或者,如果其他人已经发表了有关分子的原子分辨率的结果,你可以将这些数据作为参考材料。 溶剂的选择很重要:分子在水性溶剂或非极性溶剂中的表现可能不同,CG模型可能无法同样好地捕捉这两者。选择一种与分子将花费大部分时间的环境相当的溶剂。如果这两种环境都很重要,你可以选择妥协,两全其美。 对于甲苯,可以使用(单击要下载的文件)运行模拟: gmx grompp -f gromos.mdp -c gromos.gro -p gromos.top -o gromos gmx mdrun -v -deffnm gromos 这使您可以模拟作为溶剂的癸烷中的单个甲苯分子。请注意,该示例中使用的1ns模拟时间足够了,因为甲苯是如此简单的分子。 需要粗粒化的物质只需要一个,目的只是搜集数据,增加多了,会增加消耗的时间。
|
Powered by Discuz! X3.5
© 2001-2024 Discuz! Team.