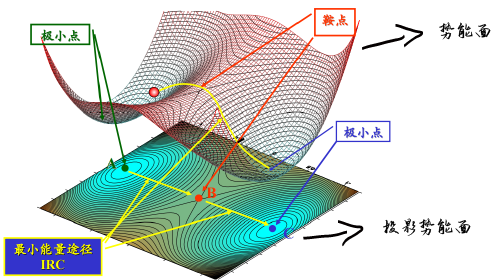
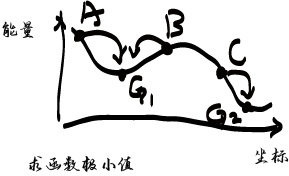| 极小点就是整体体系中能量 相对稳定的结构 对于MD来说没有必要必须找到最低值 差不多就好了,很多人喜欢算几万步 没有必要的,差不多就好了
MD中进行能量最小化的目的是 为了避免因为原子结构靠的太近 导致的斥力太大,以至于后续无法进行计算 面对部分尤其高分子类体系 需要进行做退火结构优化 单纯的通过能量最小化没有办法实现 结构达到合理状态
MD中常见的能量优化方法 一次导数求值(常用) 最速下降法SD:沿最大受力方向检索,调大步长,否则缩小步长再检索远离极小点速度快,离得近就慢 共轭梯度法CG:在SD基础上进行修正,导致前期计算速度慢但是后期精度上来,相对SD精准,速度慢 二次导数求值 牛顿拉森法Newton-Rapson:计算100个原子以内的分子还行,否则效率不好,这个里面需要计算Hessian矩阵 近似牛顿法BFGS:在牛顿拉森法中进行修正,速度慢,但是精度高,对于CG无法收敛的可以用,对于如果利用分子力学做振动相关分析可以 分子力学中,求取势能的极小值多利用导数极小化法 势能的一次导数称为梯度是向量,此向量的方向指向极小值点 其大小代表目前所处位置的倾斜度 势能函数的二次导数为势能面在此位置的弯曲度 |
Powered by Discuz! X3.5
© 2001-2024 Discuz! Team.